











Síndrome de Apert

De la mano de la Asociación Nacional del Síndrome de Apert y otras Craneosinostosis Sindrómicas, analizamos en qué consiste esta enfermedad, sus causas y tratamientos.
¿Qué es el Síndrome de Apert y las Craneosinostosis Sindrómicas?
El Síndrome de Apert es un tipo de craneosinostosis sindrómica. Esta afección craneofacial fue descrita por primera vez a principios de 1900 por el pediatra francés Eugène Apert.
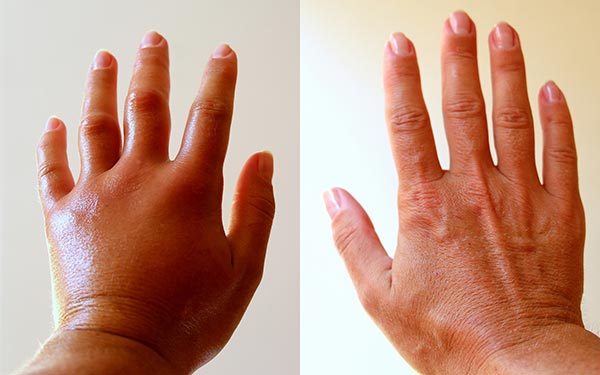
Las Craneosinostosis son defectos congénitos producidos por un cierre prematuro de una o varias de las suturas craneales que forman el cráneo. El cerebro, al continuar con su proceso natural de crecimiento, se encuentra con una barrera ósea que lo presiona, es decir, se encuentra ante un cráneo inexpandible, provocando malformaciones en la cabeza y presión intracraneal (característica común en muchos de los síndromes craneofaciales). Si también aparecen otros síntomas como la retrusión del tercio medio de la cara, alteraciones oculares y visuales, problemas auditivos, respiratorios, de alimentación, sindactilia (fusión de los dedos de las manos o los pies) o exoftalmia (ojos que sobresalen de su cavidad), y discapacidad intelectual, entre otros, entonces estaremos hablando de craneosinostosis sindrómicas.
¿Cuáles son sus características principales?
Las características más comunes que presentan las personas afectadas por estos síndromes son:
- Craneosinostosis.
- Braquicefalia (cabeza corta y ancha).
- Hipoplasia del tercio medio de la cara. Puede causar una vía respiratoria pequeña o estrecha que contribuye al aspecto de una mandíbula inferior relativamente grande y que puede dar lugar problemas respiratorios graves.
- Protrusión ocular.
- Sindactilia (fusión de los dedos de las manos y de los pies).
- Hendidura del paladar.
¿Cuál es la causa de las Craneosinostosis Sindrómicas?
Las Craneosinostosis Sindrómicas están causadas por mutaciones generadas en el periodo prenatal, en concreto, la mutación del gen del cromosoma número 10, denominado receptor 2 del factor de crecimiento de fibroblastos FGPR2. De origen desconocido, en su gran mayoría se trata de mutaciones o casos “de novo” (mutaciones espontáneas), aunque también pueden ser de origen hereditario. Su incidencia en la población general puede oscilar entre 1 y 6 casos cada 100.000 nacimientos.
¿Qué otros tipos de Craneosinostosis Sindrómicas existen y qué síntomas o características tienen?
Junto al Síndrome de Apert, existen más de 100 síndromes asociados a las Craneosinostosis Sindrómicas, siendo los más característicos:
- Síndrome de Pfeiffer: es un síndrome de dismorfología craneofacial que fue inicialmente descrito en 1964. Los niños con síndrome de Pfeiffer tienen una serie de problemas de gravedad variable, desde síntomas faciales fundamentalmente estéticos hasta síntomas graves que afectan a la respiración (la hipoplasia maxilar puede dar lugar a una laringe y a una faringe pequeñas detrás de la nariz y de la boca, restringiendo el paso del aire hacia la tráquea y los pulmones y causando dificultad respiratoria), a la alimentación, a la visión (órbitas poco profundas y exoftalmia) y al desarrollo cerebral. También se pueden dar anomalías en los dedos de las manos y de los pies.
- Síndrome de Saethre-Chotzen: fue descrito por Saethre y Chotzen a principios de la década de 1930. Los afectados tienen características muy variables, lo que dificulta el diagnóstico de nuevos casos. Los rasgos más comunes que presentan las personas afectadas por este síndrome son braquicefalia, párpados caídos, dedos de las manos cortos y dedos de los pies anchos y fusión de los huesos del cuello.
- Síndrome de Crouzon: es un síndrome de apariencia craneofacial anormal que fue inicialmente descrito en 1912. El síndrome afecta predominantemente al aspecto de la cabeza y de la cara, por lo que las personas afectadas por este síndrome presentan regresión de la cara media (dando lugar a problemas respiratorios y de alimentación) y órbitas superficiales, es decir, la cara tiene una apariencia cóncava y las órbitas poco profundas dan como resultado globos oculares prominentes, produciendo problemas de visión y problemas en el desarrollo cerebral.
- Síndrome de Muenke: el Dr. Muenke dirigió el equipo que descubrió esta enfermedad en 1996. Generalmente, este síndrome se diagnóstica debido a una craneosinostosis que afecta a las suturas coronales y, además de la distorsión que puede darse en la forma del cráneo, la apariencia es esencialmente normal. No existen unas características físicas que puedan dar el diagnóstico, como ocurre en la mayoría de los síndromes craneofaciales, únicamente se puede confirmar el diagnóstico a través de pruebas genéticas. En ocasiones, las personas afectadas pueden tener dedos de las manos cortos, ligeramente torcidos o palmeados, pero esto no afecta a su función. También puede haber una pérdida de audición asociada y dificultades de aprendizaje en el 10% -30% de los casos.
¿Tienen tratamiento las Craneosinostosis Sindrómicas?
El tratamiento de los afectados por los síndromes craneofaciales exige una intervención quirúrgica temprana, como puede ser la remodelación craneal para dar espacio al cerebro comprimido y aliviar la presión intracraneal. Posteriormente, en aquellos casos en los que sea necesario, se puede realizar el adelantamiento del tercio medio facial con avance de las orbitas para descomprimir el espacio intracraneal. De este modo, se mejorará la función respiratoria y permitiendo el desarrollo normal e impidiendo que las distintas áreas cerebrales queden afectadas. Otra opción quirúrgica consiste en la separación de los de dedos de las manos y pies.
Las intervenciones a las que pueden ser sometidas las personas afectadas pueden ser múltiples y repetirse a lo largo de toda su infancia y hasta la adolescencia tardía.
La estimulación temprana, así como la fisioterapia, terapia ocupacional, logopedia o apoyo psicológico se vuelven imprescindibles para conseguir un desarrollo equilibrado del afectado en todos los ámbitos de la vida, una mejor integración psico-social y el máximo desarrollo de sus capacidades.
Fuentes
*Esta información en ningún momento sustituye la consulta o diagnóstico de un profesional médico o farmacéutico.